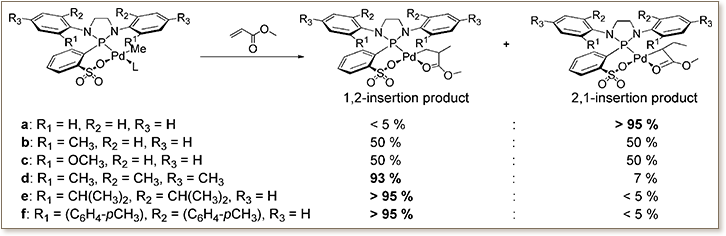
Diazaphospholidine-sulfonato Pd(II) complexes [{κ2-P,O-(N- Ar2C2H4N2P)C6H 4SO3}PdMe(L)] 1-L (L = dmso, pyridine, lutidine, or μ-LiCl(solvent); 1a: Ar = Ph, 1b: Ar = 2-MeC6H4, 1c: Ar = 2-MeOC6H4, 1d: Ar = 2,4,6-Me3C 6H2, 1e: Ar = 2,6-iPr2C6H 3, 1f: Ar = 2,6-(p-tolyl)2C6H3) were prepared and structurally characterized. The regioselectivity of methyl acrylate (MA) insertion into the Pd-Me bond is entirely inverted from >93% 1,2-insertion for bulky substituents (1d-f, yielding the insertion products [(P̂O)Pd{κ2-C,O-CH2CHMeC(O)OMe], 12) to the usual electronically controlled 2,1-insertion (>95%) for the less bulky Ar = Ph (1a, yielding the insertion product [(P̂O)Pd{κ2-C,O- CHEtC(O)OMe], 11, and β-H elimination product methyl crotonate). DFT studies underline that this is due to a more favorable insertion transition state (2,1- favored by 12 kJ mol-1 over 1,2- for 1a) vs destabilization of the 2,1-insertion transition state in 1d,e. By contrast, MA insertion into the novel isolated and structurally characterized hydride and deuteride complexes [{κ2-P,O-(N-Ar2C 2H4N2P)C6H4SO 3}PdR(lutidine)] (Ar = 2,6-iPr2C6H3; 9e: R = H, 10e: R = D) occurs 2,1-selectively. This is due to the insertion occurring from the isomer with the P-donor and the olefin in trans arrangement, rather than the insertion into the alkyl from the cis isomer in which the olefin is in proximity to the bulky diazaphospholidine. 1a-f are precursors to active catalysts for ethylene polymerization to highly linear polyethylene with M n up to 35 000 g mol-1. In copolymerization experiments, norbornene was incorporated in up to 6.1 mol % into the polyethylene backbone.
10.1021/om300755j