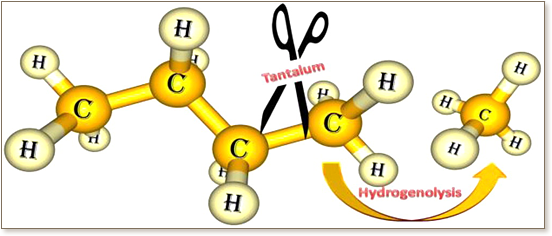
 The mechanism of hydrogenolysis of alkanes, promoted by Ta-hydrides supported on silica via 2 ≡ Si-O- bonds, has been studied with a density functional theory (DFT) approach. Our study suggests that the initial monohydride (≡ Si-O-)2Ta(III)H is rapidly trapped by molecular hydrogen to form the more stable tris-hydride (≡ Si-O-) 2Ta(V)H3. Loading of n-butane to the Ta-center occurs through C-H activation concerted with elimination of molecular hydrogen (σ-bond metathesis). Once the Ta-alkyl species is formed, the C-C activation step corresponds to a β-alkyl transfer to the metal with elimination of an olefin. According to these calculations, an α-alkyl transfer to the metal to form a Ta-carbene species is of higher energy. The olefins formed during the C-C activation step can be rapidly hydrogenated by both mono- and tris-Ta-hydride species, making the overall process of alkane cracking thermodynamically favored.
The mechanism of hydrogenolysis of alkanes, promoted by Ta-hydrides supported on silica via 2 ≡ Si-O- bonds, has been studied with a density functional theory (DFT) approach. Our study suggests that the initial monohydride (≡ Si-O-)2Ta(III)H is rapidly trapped by molecular hydrogen to form the more stable tris-hydride (≡ Si-O-) 2Ta(V)H3. Loading of n-butane to the Ta-center occurs through C-H activation concerted with elimination of molecular hydrogen (σ-bond metathesis). Once the Ta-alkyl species is formed, the C-C activation step corresponds to a β-alkyl transfer to the metal with elimination of an olefin. According to these calculations, an α-alkyl transfer to the metal to form a Ta-carbene species is of higher energy. The olefins formed during the C-C activation step can be rapidly hydrogenated by both mono- and tris-Ta-hydride species, making the overall process of alkane cracking thermodynamically favored.